MEANtools: multi-omics integration towards metabolite anticipation and biosynthetic pathway prediction (preprint)
 biorxiv
biorxivAbstract
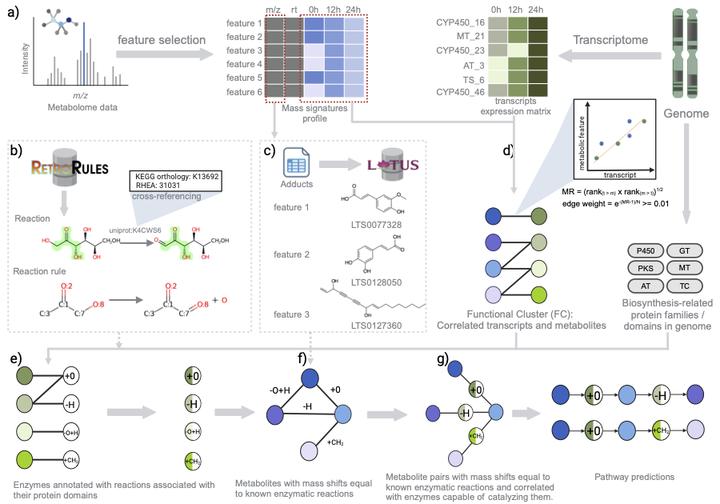
During evolution, plants have developed the ability to produce a vast array of specialized metabolites, which play crucial roles in helping plants adapt to different environmental niches. However, their biosynthetic pathways remain largely elusive. In the past decades, increasing numbers of plant biosynthetic pathways have been elucidated based on approaches utilizing genomics, transcriptomics, and metabolomics. These efforts, however, are limited by the fact that they typically adopt a target-based approach, requiring prior knowledge. Here, we present MEANtools, a systematic and unsupervised computational integrative omics workflow to predict candidate metabolic pathways de novo by leveraging knowledge of general reaction rules and metabolic structures stored in public databases. In our approach, possible connections between metabolites and transcripts that show correlated abundance across samples are identified using reaction rules linked to the transcript-encoded enzyme families. MEANtools thus assesses whether these reactions can connect transcript-correlated mass features within a candidate metabolic pathway. We validate MEANtools using a paired transcriptomic-metabolomic dataset recently generated to reconstruct the falcarindiol biosynthetic pathway in tomato. MEANtools correctly anticipated five out of seven steps of the characterized pathway and also identified other candidate pathways involved in specialized metabolism, which demonstrates its potential for hypothesis generation. Altogether, MEANtools represents a significant advancement to integrate multi-omics data for the elucidation of biochemical pathways in plants and beyond.